2025

Human clearance systems have a layered architecture across tissues and cell types that supports varied proteome compositions
Ekaterina Vinogradov-Talyah, Bar Edri , Lior Ravkaie , Or Lazarescu, Fadi Gharra , Juman Jubran, Anat Ben-Zvi , Esti Yeger-Lotem

Network-based anomaly detection algorithm reveals proteins with major roles in human tissues
Dima Kagan, Juman Jubran, Esti Yeger-Lotem, Michael Fire

Human subcutaneous and visceral adipocyte atlases uncover classical and non-classical adipocytes and depot-specific patterns
Or Lazarescu*, Maya Ziv-Agam*, Yulia Haim*, Idan Hekselman, Juman Jubran, Ariel Shneyour, Habib Muallem, Alon Zemer, Marina Rosengarten-Levin, Daniel Kitsberg, Liron Levin, Idit F Liberty, Uri Yoel, Oleg Dukhno, Miriam Adam, Julia Braune, Claudia Müller, Nora Raulien, Martin Gericke, Antje Körner, Rinki Murphy, Matthias Blüher, Naomi Habib, Assaf Rudich#, and Esti Yeger-Lotem#
2024

Tissue-aware interpretation of genetic variants advances the etiology of rare diseases
Argov CM, Shneyour A, Jubran J, Sabag E, Mansbach A, Sepunaru Y, Filtzer E, Gruber G, Volozhinsky M, Yogev Y, Birk O, Chalifa-Caspi V, Rokach L, Yeger-Lotem E#
Mol Syst Biol 2024. doi:https://doi.org/10.1038/s44320-024-00061-6
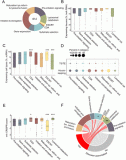
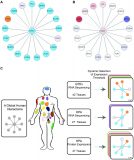
The landscape of cellular clearance systems across human tissues and cell types is shaped by tissue-specific proteome needs
Ekaterina Vinogradov, Lior Ravkaie, Bar Edri, Juman Jubran, Anat Ben-Zvi, Esti Yeger-Lotem

sNucConv: A bulk RNA-seq deconvolution method trained on single-nucleus RNA-seq data to estimate cell-type composition of human adipose tissues
Sorek G, Haim Y, Chalifa-Caspi V, Lazarescu O, Ziv-Agam M, Hagemann T, Nono Nankam PA, Blüher M, Liberty IF, Dukhno O, Kukeev I, Yeger-Lotem E, Rudich A, Levin L
iScience 2024. doi:https://doi.org/10.1016/j.isci.2024.110368

Machine-learning analysis reveals an important role for negative selection in shaping cancer aneuploidy landscapes
Juman Jubran, Rachel Slutsky, Nir Rozenblum, Lior Rokach, Uri Ben-David & Esti Yeger-Lotem
Genome Biology 2024. doi:https://doi.org/10.1186/s13059-024-03225-7

Affected cell types for hundreds of Mendelian diseases revealed by analysis of human and mouse single-cell data
Idan Hekselman, Assaf Vital, Maya Ziv-Agam, Lior Kerber, Ido Yairi, Esti Yeger-Lotem
2023
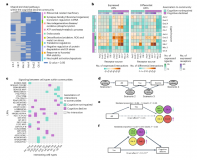
Multicellular communities are perturbed in the aging human brain and Alzheimer’s disease
Anael Cain, Mariko Taga, Cristin McCabe, Gilad S Green, Idan Hekselman, Charles C White, Dylan I Lee, Pallavi Gaur, Orit Rozenblatt-Rosen, Feng Zhang, Esti Yeger-Lotem, David A Bennett, Hyun-Sik Yang, Aviv Regev, Vilas Menon, Naomi Habib, Philip L De Jager
Nature Neuroscience 2023. doi:DOI: 10.1038/s41593-023-01356-x

Predicting molecular mechanisms of hereditary diseases by using their tissue-selective manifestation
Eyal Simonovsky, Moran Sharon, Maya Ziv, Omry Mauer, Idan Hekselman, Juman Jubran, Ekaterina Vinogradov, Chanan M Argov, Omer Basha, Lior Kerber, Yuval Yogev, Ayellet V Segrè, Hae Kyung Im, GTEx Consortium, Ohad Birk, Lior Rokach, Esti Yeger-Lotem
Molecular Systems Biology (2023) e11407 2023. doi:https://doi.org/10.15252/msb.202211407
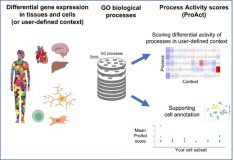
ProAct: quantifying the differential activity of biological processes in tissues, cells, and user-defined contexts
Moran Sharon, Gil Gruber, Chanan M Argov, Miri Volozhinsky, Esti Yeger-Lotem
Nucleic Acids Research 2023. doi:https://doi.org/10.1093/nar/gkad421
2022

The Organ-Disease Annotations (ODiseA) Database of Hereditary Diseases and Inflicted Tissues
Idan Hekselman, Lior Kerber, Maya Ziv, Gil Gruber, Esti Yeger-Lotem
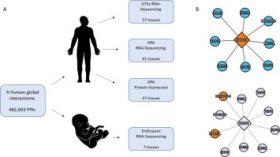
The TissueNet v.3 Database: Protein-protein Interactions in Adult and Embryonic Human Tissue contexts
Maya Ziv, Gil Gruber, Moran Sharon, Ekaterina Vinogradov, Esti Yeger-Lotem

The differential activity of biological processes in tissues and cell subsets can illuminate disease-related processes and cell type identities
Moran Sharon, Ekaterina Vinogradov, Chanan M Argov, Or Lazarescu, Yazeed Zoabi, Idan Hekselman, Esti Yeger-Lotem
Bioinformatics 2022. doi:https://doi.org/10.1093/bioinformatics/btab883
2021

Unraveling the hidden role of a uORF-encoded peptide as a kinase inhibitor of PKCs
Divya Ram Jayaram, Sigal Frost, Chanan Argov, Vijayasteltar Belsamma Liju, Nikhil Ponnoor Anto, Amitha Muraleedharan, Assaf Ben-Ari, Rose Sinay, Ilan Smoly, Ofra Novoplansky, Noah Isakov, Debra Toiber, Chen Keasar, Moshe Elkabets, Esti Yeger-Lotem, Etta Livneh
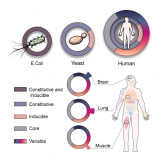
The landscape of molecular chaperones across human tissues reveals a layered architecture of core and variable chaperones
Netta Shemesh*, Juman Jubran*, Shiran Dror, Eyal Simonovsky, Omer Basha, Chanan Argov, Idan Hekselman, Mehtap Abu-Qarn, Ekaterina Vinogradov, Omry Mauer, Tatiana Tiago, Serena Carra, Anat Ben-Zvi & Esti Yeger-Lotem
Nature Communications 2021. doi:https://doi.org/10.1038/s41467-021-22369-9

A tissue-aware machine learning framework enhances the mechanistic understanding and genetic diagnosis of Mendelian and rare diseases
Eyal Simonovsky, Moran Sharon, Maya Ziv, Omry Mauer, Idan Hekselman, Juman Jubran, Ekaterina Vinogradov, Chanan M. Argov, Omer Basha, Lior Kerber, Yuval Yogev, Ayellet V. Segrè, Hae Kyung Im, GTEx Consortium, Ohad Birk, Lior Rokach, Esti Yeger-Lotem
2020
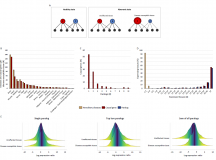
Dosage-sensitive molecular mechanisms are associated with the tissue-specificity of traits and diseases
Juman Jubran, Idan Hekselman, Lena Novack, Esti Yeger-Lotem
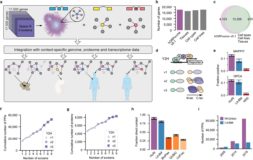
A reference map of the human protein interactome
Katja Luck, Dae-Kyum Kim, Luke Lambourne, Kerstin Spirohn, Bridget E. Begg, Wenting Bian, Ruth Brignall, Tiziana Cafarelli, Francisco J. Campos-Laborie, Benoit Charloteaux, Dongsic Choi, Atina G. Cote, Meaghan Daley, Steven Deimling, Alice Desbuleux, Amélie Dricot, Marinella Gebbia, Madeleine F. Hardy, Nishka Kishore, Jennifer J. Knapp, István A. Kovács, Irma Lemmens, Miles W. Mee, Joseph C. Mellor, Carl Pollis, Carles Pons, Aaron D. Richardson, Sadie Schlabach, Bridget Teeking, Anupama Yadav, Mariana Babor, Dawit Balcha, Omer Basha, Suet-Feung Chin, Soon Gang Choi, Claudia Colabella, Georges Coppin, Cassandra D’Amata, David De Ridder, Steffi De Rouck, Miquel Duran-Frigola, Hanane Ennajdaoui, Florian Goebels, Anjali Gopal, Ghazal Haddad, Mohamed Helmy, Yves Jacob, Yoseph Kassa, Roujia Li, Natascha van Lieshout, Andrew MacWilliams, Dylan Markey, Joseph N. Paulson, Sudharshan Rangarajan, John Rasla, Ashyad Rayhan, Thomas Rolland, Adriana San Miguel, Yun Shen, Dayag Sheykhkarimli, Gloria M. Sheynkman, Eyal Simonovsky, Murat Taşan, Alexander Tejeda, Jean-Claude Twizere, Yang Wang, Robert Weatheritt, Jochen Weile, Yu Xia, Xinping Yang, Esti Yeger-Lotem, Quan Zhong, Patrick Aloy, Gary D. Bader, Javier De Las Rivas, Suzanne Gaudet, Tong Hao, Janusz Rak, Jan Tavernier, Vincent Tropepe, David E. Hill, Marc Vidal, Frederick P. Roth, Michael A. Calderwood
Nature 2020 580(7803):402-408. doi:10.1038/s41586-020-2188-x.
Global insights into cellular organization and function require comprehensive understanding of interactome networks. Similar to how a reference genome sequence recently revolutionized human genetics, a reference map of the human interactome network is critical to fully understand genotype-phenotype relationships. Here, we present the first human “all-by-all” binary reference interactome map, or “HuRI”. With ∼53,000 high-quality protein-protein interactions (PPIs), HuRI is approximately four times larger than the information curated from small-scale studies available in the literature. Integrating HuRI with genome, transcriptome and proteome data enables the study of cellular function within essentially any physiological or pathological cellular context. We demonstrate the use of HuRI in identifying specific subcellular roles of PPIs and protein function modulation via splicing during brain development. Inferred tissue-specific networks reveal general principles for the formation of cellular context-specific functions and elucidate potential molecular mechanisms underlying tissue-specific phenotypes of Mendelian diseases. HuRI thus represents an unprecedented, systematic reference linking genomic variation to phenotypic outcomes.
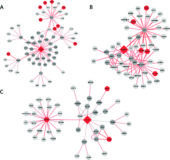
Differential network analysis of human tissue interactomes highlights tissue-selective processes and genetic disorder genes
Omer Basha*, Chanan M Argov*, Raviv Artzy, Yazeed Zoabi, Idan Hekselman, Liad Alfandari, Vered Chlifa-Caspi and Esti Yeger-Lotem
Bioinformatics. 2020 pii: btaa034. doi:10.1093/bioinformatics/btaa034.
MOTIVATION: Differential network analysis, designed to highlight network changes between conditions, is an important paradigm in network biology. However, differential network analysis methods have been typically designed to compare between two conditions, and were rarely applied to multiple protein interaction networks (interactomes). Importantly, large-scale benchmarks for their evaluation have been lacking.
RESULTS: Here, we present a framework for assessing the ability of differential network analysis of multiple human tissue interactomes to highlight tissue-selective processes and disorders. For this, we created a benchmark of 6,499 curated tissue-specific Gene Ontology biological processes. We applied five methods, including four differential network analysis methods, to construct weighted interactomes for 34 tissues. Rigorous assessment of this benchmark revealed that differential analysis methods perform well in revealing tissue-selective processes (AUCs of 0.82-0.9). Next, we applied differential network analysis to illuminate the genes underlying tissue-selective hereditary disorders. For this, we curated a dataset of 1,305 tissue-specific hereditary disorders and their manifesting tissues. Focusing on subnetworks containing the top 1% differential interactions in disease-relevant tissue interactomes revealed significant enrichment for disorder-causing genes in 18.6% of the cases, with a significantly high success rate for blood, nerve, muscle and heart diseases.
SUMMARY: We offer a framework that includes expansive manually-curated datasets of tissue-selective processes and disorders to be used as benchmarks or to illuminate tissue-selective processes and genes. Our results demonstrate that differential analysis of multiple human tissue interactomes is a powerful tool for highlighting processes and genes with tissue-selective functionality and clinical impact.

Mechanisms of tissue and cell-type specificity in heritable traits and diseases
Idan Hekselman & Esti Yeger-Lotem
Hundreds of heritable traits and diseases that are caused by germlineaberrations in ubiquitously expressed genes manifest in a remarkably limited number of cell types and tissues across the body. Unravelling mechanisms that govern their tissue-specific manifestations is critical for our understanding of disease aetiologies and may direct efforts to develop treatments. Owing to recent advances in high-throughput technologies and open resources, data and tools are now available to approach this enigmatic phenomenon at large scales, both computationally and experimentally. Here, we discuss the large prevalence of tissue-selective traits and diseases, describe common molecular mechanisms underlying their tissue-selective manifestation and present computational strategies and publicly available resources for elucidating the molecular basis of their genotype–phenotype relationships.
2019

Aging promotes reorganization of the CD4 T cell landscape toward extreme regulatory and effector phenotypes.
Yehezqel Elyahu*, Idan Hekselman*, Inbal Eizenberg-Magar, Omer Berner, Itai Strominger, Maya Schiller, Kritika Mittal, Anna Nemirovsky, Ekaterina Eremenko, Assaf Vital, Eyal Simonovsky, Vered Chalifa-Caspi, Nir Friedman, Esti Yeger-Lotem, and Alon Monsonego
Science Advances 2019 5:8. doi:10.1126/sciadv.aaw8330
Age-associated changes in CD4 T-cell functionality have been linked to chronic inflammation and decreased immunity. However, a detailed characterization of CD4 T cell phenotypes that could explain these dysregulated functional properties is lacking. We used single-cell RNA sequencing and multidimensional protein analyses to profile thousands of CD4 T cells obtained from young and old mice. We found that the landscape of CD4 T cell subsets differs markedly between young and old mice, such that three cell subsets—exhausted, cytotoxic, and activated regulatory T cells (aTregs)—appear rarely in young mice but gradually accumulate with age. Most unexpected were the extreme pro- and anti-inflammatory phenotypes of cytotoxic CD4 T cells and aTregs, respectively. These findings provide a comprehensive view of the dynamic reorganization of the CD4 T cell milieu with age and illuminate dominant subsets associated with chronic inflammation and immunity decline, suggesting new therapeutic avenues for age-related diseases.
ResponseNet v.3: Revealing signaling and regulatory pathways connecting your proteins and genes across human tissues
Omer Basha, Omry Mauer, Eyal Simonovsky, Rotem Shpringer and Esti Yeger-Lotem
Nucleic Acids Research, In Press 2019. doi:https://doi.org/10.1093/nar/gkz421
ResponseNet v.3 is an enhanced version of ResponseNet, a web server that is designed to highlight signaling and regulatory pathways connecting user-defined proteins and genes by using the ResponseNet network optimization approach (http://netbio.bgu.ac.il/respnet). Users run ResponseNet by defining source and target sets of proteins, genes and/or microRNAs, and by specifying a molecular interaction network (interactome). The output of ResponseNet is a sparse, high-probability interactome subnetwork that connects the two sets, thereby revealing additional molecules and interactions that are involved in the studied condition. In recent years, massive efforts were invested in profiling the transcriptomes of human tissues, enabling the inference of human tissue interactomes. ResponseNet v.3 expands ResponseNet2.0 by harnessing ∼11,600 RNA-sequenced human tissue profiles made available by the Genotype-Tissue Expression consortium, to support context-specific analysis of 44 human tissues. Thus, ResponseNet v.3 allows users to illuminate the signaling and regulatory pathways potentially active in the context of a specific tissue, and to compare them with active pathways in other tissues. In the era of precision medicine, such analyses open the door for tissue- and patient-specific analyses of pathways and diseases.
Elucidating genotype-to-phenotype relationships via analyses of human tissue interactomes
Idan Hekselman, Moran Sharon, Omer Basha and Esti Yeger-Lotem

Large-scale analysis of human gene expression variability associates highly variable drug targets with lower drug effectiveness and safety
Eyal Simonovsky, Ronen Schuster and Esti Yeger-Lotem
Bioinformatics 2019 5(17):3028-3037. doi:https://doi.org/10.1093/bioinformatics/btz023
Motivation
Results
To explore this hypothesis, we analyzed the expression variability of protein-coding genes, and particularly drug target genes, across individuals. For this, we developed a novel variability measure, termed local coefficient of variation (LCV), which ranks the expression variability of each gene relative to genes with similar expression levels. Unlike commonly used methods, LCV neutralizes expression levels biases without imposing any distribution over the variation and is robust to data incompleteness. Application of LCV to RNA-sequencing profiles of 19 human tissues and to target genes of 1076 approved drugs revealed that drug target genes were significantly more variable than protein-coding genes. Analysis of 113 drugs with available effectiveness scores showed that drugs targeting highly variable genes tended to be less effective in the population. Furthermore, comparison of approved drugs to drugs that were withdrawn from the market showed that withdrawn drugs targeted significantly more variable genes than approved drugs. Last, upon analyzing gender differences we found that the variability of drug target genes was similar between men and women. Altogether, our results suggest that expression variability of drug target genes could contribute to the variable responsiveness and effectiveness of drugs, and is worth considering during drug treatment and development.
Availability and implementation
LCV is available as a python script in GitHub (https://github.com/eyalsim/LCV).
2018

Integrating Rio1 activities discloses its nutrient-activated network in Saccharomyces cerevisiae
Maria G Iacovella, Michael Bremang, Omer Basha, Luciano Giacò, Walter Carotenuto, Cristina Golfieri, Barnabas Szakal, Marianna Dal Maschio, Valentina Infantino, Galina V Beznoussenko, Chinnu R Joseph, Clara Visintin, Alexander A Mironov, Rosella Visintin, Dana Branzei, Sébastien Ferreira-Cerca, Esti Yeger-Lotem, Peter De Wulf
Nucleic Acids Research 2018. doi:https://doi.org/10.1093/nar/gky618
The Saccharomyces cerevisiae kinase/adenosine triphosphatase Rio1 regulates rDNA transcription and segregation, pre-rRNA processing and small ribosomal subunit maturation. Other roles are unknown. When overexpressed, human ortholog RIOK1 drives tumor growth and metastasis. Likewise, RIOK1 promotes 40S ribosomal subunit biogenesis and has not been characterized globally. We show that Rio1 manages directly and via a series of regulators, an essential signaling network at the protein, chromatin and RNA levels. Rio1 orchestrates growth and division depending on resource availability, in parallel to the nutrient-activated Tor1 kinase. To define the Rio1 network, we identified its physical interactors, profiled its target genes/transcripts, mapped its chromatin-binding sites and integrated our data with yeast’s protein–protein and protein–DNA interaction catalogs using network computation. We experimentally confirmed network components and localized Rio1 also to mitochondria and vacuoles. Via its network, Rio1 commands protein synthesis (ribosomal gene expression, assembly and activity) and turnover (26S proteasome expression), and impinges on metabolic, energy-production and cell-cycle programs. We find that Rio1 activity is conserved to humans and propose that pathological RIOK1 may fuel promiscuous transcription, ribosome production, chromosomal instability, unrestrained metabolism and proliferation; established contributors to cancer. Our study will advance the understanding of numerous processes, here revealed to depend on Rio1 activity.
Role of duplicate genes in determining the tissue-selectivity of hereditary diseases
Ruth Barshir, Idan Hekselman, Netta Shemesh, Moran Sharon, Lena Novack, Esti Yeger-Lotem
PLoS Genetics 2018. doi:https://doi.org/10.1371/journal.pgen.1007327
A longstanding puzzle in human genetics is what limits the clinical manifestation of hundreds of hereditary diseases to certain tissues, while their causal genes are expressed throughout the human body. A general conception is that tissue-selective disease phenotypes emerge when masking factors operate in unaffected tissues, but are specifically absent or insufficient in disease-manifesting tissues. Although this conception has critical impact on the understanding of disease manifestation, it was never challenged in a systematic manner across a variety of hereditary diseases and affected tissues. Here, we address this gap in our understanding via rigorous analysis of the susceptibility of over 30 tissues to 112 tissue-selective hereditary diseases. We focused on the roles of paralogs of causal genes, which are presumably capable of compensating for their aberration. We show for the first time at large-scale via quantitative analysis of omics datasets that, preferentially in the disease-manifesting tissues, paralogs are under-expressed relative to causal genes in more than half of the diseases. This was observed for several susceptible tissues and for causal genes with varying number of paralogs, suggesting that imbalanced expression of paralogs increases tissue susceptibility. While for many diseases this imbalance stemmed from up-regulation of the causal gene in the disease-manifesting tissue relative to other tissues, it was often combined with down-regulation of its paralog. Notably in roughly 20% of the cases, this imbalance stemmed only from significant down-regulation of the paralog. Thus, dosage relationships between paralogs appear as important, yet currently under-appreciated, modifiers of disease manifestation.
RSRC1 mutation affects intellect and behaviour through aberrant splicing and transcription, downregulating IGFBP3
Yonatan Perez, Shay Menascu, Idan Cohen, Rotem Kadir, Omer Basha, Zamir Shorer, Hila Romi, Gal Meiri, Tatiana Rabinski, Rivka Ofir, Esti Yeger-Lotem, Ohad S Birk
Brain 2018 141:4:961-970. doi:https://doi.org/10.1093/brain/awy045
RSRC1, whose polymorphism is associated with altered brain function in schizophrenia, is a member of the serine and arginine rich-related protein family. Through homozygosity mapping and whole exome sequencing we show that RSRC1 mutation causes an autosomal recessive syndrome of intellectual disability, aberrant behaviour, hypotonia and mild facial dysmorphism with normal brain MRI. Further, we show that RSRC1 is ubiquitously expressed, and that the RSRC1 mutation triggers nonsense-mediated mRNA decay of the RSRC1transcript in patients’ fibroblasts. Short hairpin RNA (shRNA)-mediated lentiviral silencing and overexpression of RSRC1 in SH-SY5Y cells demonstrated that RSRC1 has a role in alternative splicing and transcription regulation. Transcriptome profiling of RSRC1-silenced cells unravelled specific differentially expressed genes previously associated with intellectual disability, hypotonia and schizophrenia, relevant to the disease phenotype. Protein-protein interaction network modelling suggested possible intermediate interactions by which RSRC1 affects gene-specific differential expression. Patient-derived induced pluripotent stem cells, differentiated into neural progenitor cells, showed expression dynamics similar to the RSRC1-silenced SH-SY5Y model. Notably, patient neural progenitor cells had 9.6-fold downregulated expression of IGFBP3, whose brain expression is affected by MECP2, aberrant in Rett syndrome. Interestingly, Igfbp3-null mice have behavioural impairment, abnormal synaptic function and monoaminergic neurotransmission, likely correlating with the disease phenotype.
The DifferentialNet database of differential protein–protein interactions in human tissues
Omer Basha, Rotem Shpringer, Chanan M Argov and Esti Yeger-Lotem
Nucleic Acids Research 2018 46:D1:D522-D526. doi:https://doi.org/10.1093/nar/gkx981
DifferentialNet is a novel database that provides users with differential interactome analysis of human tissues (http://netbio.bgu.ac.il/diffnet/). Users query DifferentialNet by protein, and retrieve its differential protein–protein interactions (PPIs) per tissue via an interactive graphical interface. To compute differential PPIs, we integrated available data of experimentally detected PPIs with RNA-sequencing profiles of tens of human tissues gathered by the Genotype-Tissue Expression consortium (GTEx) and by the Human Protein Atlas (HPA). We associated each PPI with a score that reflects whether its corresponding genes were expressed similarly across tissues, or were up- or down-regulated in the selected tissue. By this, users can identify tissue-specific interactions, filter out PPIs that are relatively stable across tissues, and highlight PPIs that show relative changes across tissues. The differential PPIs can be used to identify tissue-specific processes and to decipher tissue-specific phenotypes. Moreover, they unravel processes that are tissue-wide yet tailored to the specific demands of each tissue.
2017

Genetic effects on gene expression across human tissues
GTEx Consortium
Nature 2017 550:204-213. doi:https://doi.org/10.1038/nature24277
Characterization of the molecular function of the human genome and its variation across individuals is essential for identifying the cellular mechanisms that underlie human genetic traits and diseases. The Genotype-Tissue Expression (GTEx) project aims to characterize variation in gene expression levels across individuals and diverse tissues of the human body, many of which are not easily accessible. Here we describe genetic effects on gene expression levels across 44 human tissues. We find that local genetic variation affects gene expression levels for the majority of genes, and we further identify inter-chromosomal genetic effects for 93 genes and 112 loci. On the basis of the identified genetic effects, we characterize patterns of tissue specificity, compare local and distal effects, and evaluate the functional properties of the genetic effects. We also demonstrate that multi-tissue, multi-individual data can be used to identify genes and pathways affected by human disease-associated variation, enabling a mechanistic interpretation of gene regulation and the genetic basis of disease.
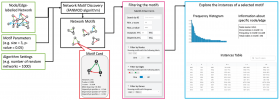
MotifNet: a web-server for network motif analysis
Ilan Y Smoly, Eugene Lerman, Michal Ziv-Ukelson, Esti Yeger-Lotem
Bioinformatics 2017 33:12:1907-1909. doi:https://doi.org/10.1093/bioinformatics/btx056
Network motifs are small topological patterns that recur in a network significantly more often than expected by chance. Their identification emerged as a powerful approach for uncovering the design principles underlying complex networks. However, available tools for network motif analysis typically require download and execution of computationally intensive software on a local computer. We present MotifNet, the first open-access web-server for network motif analysis. MotifNet allows researchers to analyze integrated networks, where nodes and edges may be labeled, and to search for motifs of up to eight nodes. The output motifs are presented graphically and the user can interactively filter them by their significance, number of instances, node and edge labels, and node identities, and view their instances. MotifNet also allows the user to distinguish between motifs that are centered on specific nodes and motifs that recur in distinct parts of the network.

An Asymmetrically Balanced Organization of Kinases versus Phosphatases across Eukaryotes Determines Their Distinct Impacts
Ilan Smoly, Netta Shemesh, Michal Ziv-Ukelson, Anat Ben-Zvi and Esti Yeger-Lotem
PLoS Comp. Biol. 2017. doi:https://doi.org/10.1371/journal.pcbi.1005221
Protein phosphorylation underlies cellular response pathways across eukaryotes and is governed by the opposing actions of phosphorylating kinases and de-phosphorylating phosphatases. While kinases and phosphatases have been extensively studied, their organization and the mechanisms by which they balance each other are not well understood. To address these questions we performed quantitative analyses of large-scale ‘omics’ datasets from yeast, fly, plant, mouse and human. We uncovered an asymmetric balance of a previously-hidden scale: Each organism contained many different kinase genes, and these were balanced by a small set of highly abundant phosphatase proteins. Kinases were much more responsive to perturbations at the gene and protein levels. In addition, kinases had diverse scales of phenotypic impact when manipulated. Phosphatases, in contrast, were stable, highly robust and flatly organized, with rather uniform impact downstream. We validated aspects of this organization experimentally in nematode, and supported additional aspects by theoretic analysis of the dynamics of protein phosphorylation. Our analyses explain the empirical bias in the protein phosphorylation field toward characterization and therapeutic targeting of kinases at the expense of phosphatases. We show quantitatively and broadly that this is not only a historical bias, but stems from wide-ranging differences in their organization and impact. The asymmetric balance between these opposing regulators of protein phosphorylation is also common to opposing regulators of two other post-translational modification systems, suggesting its fundamental value.
The TissueNet v.2 database: A quantitative view of proteins and their interactions across human tissues
Basha O, Barshir R, Sharon M, Lerman E, Kirson B, Hekselman I and Yeger-Lotem E
Nucleic Acids Research 2017 45:D1:D427-D431
Knowledge of the molecular interactions of human proteins within tissues is important for identifying their tissue-specific roles and for shedding light on tissue phenotypes. However, many protein-protein interactions (PPIs) have no tissue-contexts. The TissueNet database bridges this gap by associating experimentally-identified PPIs with human tissues that were shown to express both pair-mates. Users can select a protein and a tissue, and obtain a network view of the query protein and its tissue-associated PPIs. TissueNet v.2 is an updated version of the TissueNet database previously featured in NAR. It includes over 40 human tissues profiled via RNA-sequencing or protein-based assays. Users can select their preferred expression data source and interactively set the expression threshold for determining tissue-association. The output of TissueNet v.2 underlines qualitative and quantitative features of query proteins and their PPIs. The tissue-specificity view highlights tissue-specific and globally-expressed proteins, and the quantitative view highlights proteins that were differentially expressed in the selected tissue relative to all other tissues. Together, these views allow users to quickly assess the unique versus global functionality of query proteins. Thus, TissueNet v.2 offers an extensive, quantitative and user-friendly interface to study the roles of human proteins across tissues. TissueNet v.2 is available at http://netbio.bgu.ac.il/tissuenet.
2016

A Differentiation Transcription Factor Establishes Muscle-Specific Proteostasis in Caenorhabditis elegans
Yael Bar-Lavan, Netta Shemesh, Shiran Dror, Rivka Ofir, Esti Yeger-Lotem, Anat Ben-Zvi
PLoS Genetics 2016. doi:https://doi.org/10.1371/journal.pgen.1006531
Safeguarding the proteome is central to the health of the cell. In multi-cellular organisms, the composition of the proteome, and by extension, protein-folding requirements, varies between cells. In agreement, chaperone network composition differs between tissues. Here, we ask how chaperone expression is regulated in a cell type-specific manner and whether cellular differentiation affects chaperone expression. Our bioinformatics analyses show that the myogenic transcription factor HLH-1 (MyoD) can bind to the promoters of chaperone genes expressed or required for the folding of muscle proteins. To test this experimentally, we employed HLH-1 myogenic potential to genetically modulate cellular differentiation of Caenorhabditis elegans embryonic cells by ectopically expressing HLH-1 in all cells of the embryo and monitoring chaperone expression. We found that HLH-1-dependent myogenic conversion specifically induced the expression of putative HLH-1-regulated chaperones in differentiating muscle cells. Moreover, disrupting the putative HLH-1-binding sites on ubiquitously expressed daf-21(Hsp90) and muscle-enriched hsp-12.2(sHsp) promoters abolished their myogenic-dependent expression. Disrupting HLH-1 function in muscle cells reduced the expression of putative HLH-1-regulated chaperones and compromised muscle proteostasis during and after embryogenesis. In turn, we found that modulating the expression of muscle chaperones disrupted the folding and assembly of muscle proteins and thus, myogenesis. Moreover, muscle-specific over-expression of the DNAJB6 homolog DNJ-24, a limb-girdle muscular dystrophy-associated chaperone, disrupted the muscle chaperone network and exposed synthetic motility defects. We propose that cellular differentiation could establish a proteostasis network dedicated to the folding and maintenance of the muscle proteome. Such cell-specific proteostasis networks can explain the selective vulnerability that many diseases of protein misfolding exhibit even when the misfolded protein is ubiquitously expressed.
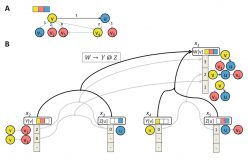
Algorithms for Regular Tree Grammar Network Search and Their Application to Mining Human-Viral Infection Patterns
Ilan Smoly, Amir Carmel, Yonat Shemer-Avni, Esti Yeger-Lotem and Michal Ziv-Ukelson
J Comput Biol. 2016 23:3:165-179. doi:https://doi.org/10.1089/cmb.2015.0168
Network querying is a powerful approach to mine molecular interaction networks. Most state-of-the-art network querying tools either confine the search to a prespecified topology in the form of some template subnetwork, or do not specify any topological constraints at all. Another approach is grammar-based queries, which are more flexible and expressive as they allow for expressing the topology of the sought pattern according to some grammar-based logic. Previous grammar-based network querying tools were confined to the identification of paths. In this article, we extend the patterns identified by grammar-based query approaches from paths to trees. For this, we adopt a higher order query descriptor in the form of a regular tree grammar (RTG). We introduce a novel problem and propose an algorithm to search a given graph for the khighest scoring subgraphs matching a tree accepted by an RTG. Our algorithm is based on the combination of dynamic programming with color coding, and includes an extension of previous k-best parsing optimization approaches to avoid isomorphic trees in the output. We implement the new algorithm and exemplify its application to mining viral infection patterns within molecular interaction networks. Our code is available online.
2015
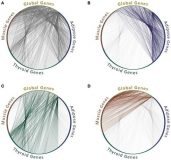
Human protein interaction networks across tissues and diseases
Esti Yeger-Lotem and Roded Sharan
Frontiers in Genetics 2015 6. doi:https://doi.org/10.3389/fgene.2015.00257
Protein interaction networks are an important framework for studying protein function, cellular processes, and genotype-to-phenotype relationships. While our view of the human interaction network is constantly expanding, less is known about networks that form in biologically important contexts such as within distinct tissues or in disease conditions. Here we review efforts to characterize these networks and to harness them to gain insights into the molecular mechanisms underlying human disease.

MyProteinNet: Build up-to-date protein interaction networks for organisms, tissues and user-defined contexts
Omer Basha, Dvir Flom, Ruth Barshir, Ilan Smoly, Shoval Tirman & Esti Yeger-Lotem
Nucleic Acids Research 2015 43:W1:W258-W263. doi:10.1093/nar/gkv515
The identification of the molecular pathways active in specific contexts, such as disease states or drug responses, often requires an extensive view of the potential interactions between a subset of proteins. This view is not easily obtained: it requires the integration of context-specific protein list or expression data with up-to-date data of protein interactions that are typically spread across multiple databases. The MyProteinNet web-server allows users to easily create such context-sensitive protein interaction networks. Users can automatically gather and consolidate data from up to 11 different databases to create a generic protein interaction network (interactome). They can score the interactions based on reliability, and filter them by user-defined contexts including molecular expression and protein annotation. The output of MyProteinNet includes the generic and filtered interactome files, together with a summary of their network attributes. MyProteinNet is particularly geared toward building human tissue interactomes, by maintaining tissue expression profiles from multiple resources. The ability of MyProteinNet to facilitate the construction of up-to-date, context-specific interactomes, and its applicability to 11 different organisms and to tens of human tissues, make it a powerful tool in meaningful analysis of protein networks.

A combination of gene expression ranking and co-expression network analysis increases discovery rate in large-scale mutant screens for novel Arabidopsis thaliana abiotic stress genes.
Vanessa Ransbotyn, Esti Yeger-Lotem, Omer Basha, Tania Acuna, Christoph Verduyn, Michal Gordon, Vered Chalifa-Caspi, Matthew A. Hannah and Simon Barak
Plant Biotechnol J. 2015 13:4:501-513. doi:10.1111/pbi.12274
As challenges to food security increase, the demand for lead genes for improving crop production is growing. However, genetic screens of plant mutants typically yield very low frequencies of desired phenotypes. Here, we present a powerful computational approach for selecting candidate genes for screening insertion mutants. We combined ranking of Arabidopsis thaliana regulatory genes according to their expression in response to multiple abiotic stresses (Multiple Stress [MST] score), with stress-responsive RNA co-expression network analysis to select candidate multiple stress regulatory (MSTR) genes. Screening of 62 T-DNA insertion mutants defective in candidate MSTR genes, for abiotic stress germination phenotypes yielded a remarkable hit rate of up to 62%; this gene discovery rate is 48-fold greater than that of other large-scale insertional mutant screens. Moreover, the MST score of these genes could be used to prioritize them for screening. To evaluate the contribution of the co-expression analysis, we screened 64 additional mutant lines of MST-scored genes that did not appear in the RNA co-expression network. The screening of these MST-scored genes yielded a gene discovery rate of 36%, which is much higher than that of classic mutant screens but not as high as when picking candidate genes from the co-expression network. The MSTR co-expression network that we created, AraSTressRegNet is publicly available at http://netbio.bgu.ac.il/arnet. This systems biology-based screening approach combining gene ranking and network analysis could be generally applicable to enhancing identification of genes regulating additional processes in plants and other organisms provided that suitable transcriptome data are available.
2014
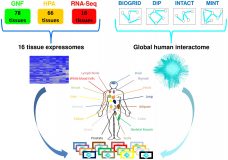
Comparative Analysis of Human Tissue Interactomes Reveals Factors Leading to Tissue-Specific Manifestation of Hereditary Diseases.
Barshir R, Shwartz O, Smoly IY & Yeger-Lotem E.
PLoS Comput Biol 2014 10:6:e1003632. doi:https://doi.org/10.1371/journal.pcbi.1003632
An open question in human genetics is what underlies the tissue-specific manifestation of hereditary diseases, which are caused by genomic aberrations that are present in cells across the human body. Here we analyzed this phenomenon for over 300 hereditary diseases by using comparative network analysis. We created an extensive resource of protein expression and interactions in 16 main human tissues, by integrating recent data of gene and protein expression across tissues with data of protein-protein interactions (PPIs). The resulting tissue interaction networks (interactomes) shared a large fraction of their proteins and PPIs, and only a small fraction of them were tissue-specific. Applying this resource to hereditary diseases, we first show that most of the disease-causing genes are widely expressed across tissues, yet, enigmatically, cause disease phenotypes in few tissues only. Upon testing for factors that could lead to tissue-specific vulnerability, we find that disease-causing genes tend to have elevated transcript levels and increased number of tissue-specific PPIs in their disease tissues compared to unaffected tissues. We demonstrate through several examples that these tissue-specific PPIs can highlight disease mechanisms, and thus, owing to their small number, provide a powerful filter for interrogating disease etiologies. As two thirds of the hereditary diseases are associated with these factors, comparative tissue analysis offers a meaningful and efficient framework for enhancing the understanding of the molecular basis of hereditary diseases.

Cancer evolution is associated with pervasive positive selection on globally expressed genes.
Ostrow SL, Barshir R, DeGregori J, Yeger-Lotem E & Hershberg R.
PLoS Genet. 2014 10:3:e1004239. doi:https://doi.org/10.1371/journal.pgen.1004239
Cancer is an evolutionary process in which cells acquire new transformative, proliferative and metastatic capabilities. A full understanding of cancer requires learning the dynamics of the cancer evolutionary process. We present here a large-scale analysis of the dynamics of this evolutionary process within tumors, with a focus on breast cancer. We show that the cancer evolutionary process differs greatly from organismal (germline) evolution. Organismal evolution is dominated by purifying selection (that removes mutations that are harmful to fitness). In contrast, in the cancer evolutionary process the dominance of purifying selection is much reduced, allowing for a much easier detection of the signals of positive selection (adaptation). We further show that, as a group, genes that are globally expressed across human tissues show a very strong signal of positive selection within tumors. Indeed, known cancer genes are enriched for global expression patterns. Yet, positive selection is prevalent even on globally expressed genes that have not yet been associated with cancer, suggesting that globally expressed genes are enriched for yet undiscovered cancer related functions. We find that the increased positive selection on globally expressed genes within tumors is not due to their expression in the tissue relevant to the cancer. Rather, such increased adaptation is likely due to globally expressed genes being enriched in important housekeeping and essential functions. Thus, our results suggest that tumor adaptation is most often mediated through somatic changes to those genes that are important for the most basic cellular functions. Together, our analysis reveals the uniqueness of the cancer evolutionary process and the particular importance of globally expressed genes in driving cancer initiation and progression.
2013
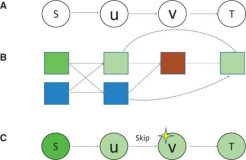
A context-sensitive framework for the analysis of human signalling pathways in molecular interaction networks
Lan A, Ziv-Ukelson M, Yeger-Lotem E
Bioinformatics 2013 29:13:i210-i216. doi:https://doi.org/10.1093/bioinformatics/btt240
Motivation: A major challenge in systems biology is to reveal the cellular pathways that give rise to specific phenotypes and behaviours. Current techniques often rely on a network representation of molecular interactions, where each node represents a protein or a gene and each interaction is assigned a single static score. However, the use of single interaction scores fails to capture the tendency of proteins to favour different partners under distinct cellular conditions.
Results: Here, we propose a novel context-sensitive network model, in which genes and protein nodes are assigned multiple contexts based on their gene ontology annotations, and their interactions are associated with multiple context-sensitive scores. Using this model, we developed a new approach and a corresponding tool, ContextNet, based on a dynamic programming algorithm for identifying signalling paths linking proteins to their downstream target genes. ContextNet finds high-ranking context-sensitive paths in the interactome, thereby revealing the intermediate proteins in the path and their path-specific contexts. We validated the model using 18 348 manually curated cellular paths derived from the SPIKE database. We next applied our framework to elucidate the responses of human primary lung cells to influenza infection. Top-ranking paths were much more likely to contain infection-related proteins, and this likelihood was highly correlated with path score. Moreover, the contexts assigned by the algorithm pointed to putative, as well as previously known responses to viral infection. Thus, context sensitivity is an important extension to current network biology models and can be efficiently used to elucidate cellular response mechanisms.

ResponseNet2.0: revealing signaling and regulatory pathways connecting your proteins and genes–now with human data.
Omer Basha, Shoval Tirman, Amir Eluk and Esti Yeger-Lotem
Nucleic Acids Research 2013 41:W1:W198-W203
Genome sequencing and transcriptomic profiling are two widely-used approaches for the identification of human disease pathways. However, each approach typically provides a limited view of disease pathways: Genome sequencing can identify disease-related mutations but rarely reveals their mode-of-action, while transcriptomic assays do not reveal the series of events that lead to the transcriptomic change. ResponseNet is an integrative network-optimization approach that we developed to fill these gaps by highlighting major signaling and regulatory molecular interaction paths that connect disease-related mutations and genes. The ResponseNet web-server provides a user-friendly interface to ResponseNet. Specifically, users can upload weighted lists of proteins and genes and obtain a sparse, weighted, molecular interaction sub-network connecting them that is biased toward regulatory and signaling pathways. ResponseNet2.0 enhances the functionality of the ResponseNet web-server in two important ways. First, it supports analysis of human data by offering a human interactome composed of proteins, genes and micro-RNAs. Second, it offers a new, informative view of the output, including a randomization analysis, in order to help users assess the biological relevance of the output sub-network. ResponseNet2.0 is available at http://netbio.bgu.ac.il/respnet
The TissueNet database of human tissue protein-protein interactions
Barshir R, Basha O, Eluk A, Smoly IY, Lan A, Yeger-Lotem E.
Nucleic Acids Research 2013 41:D1:D841-D844
Knowledge of protein–protein interactions (PPIs) is important for identifying the functions of proteins and the processes they are involved in. Although data of human PPIs are easily accessible through several public databases, these databases do not specify the human tissues in which these PPIs take place. The TissueNet database of human tissue PPIs (http://netbio.bgu.ac.il/tissuenet/) associates each interaction with human tissues that express both pair mates. This was achieved by integrating current data of experimentally detected PPIs with extensive data of gene and protein expression across 16 main human tissues. Users can query TissueNet using a protein and retrieve its PPI partners per tissue, or using a PPI and retrieve the tissues expressing both pair mates. The graphical representation of the output highlights tissue-specific and tissue-wide PPIs. Thus, TissueNet provides a unique platform for assessing the roles of human proteins and their interactions across tissues.
2011
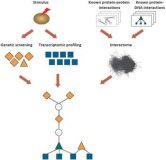
ResponseNet: revealing signaling and regulatory networks linking genetic and transcriptomic screening data
Lan A, Smoly IY, Rapaport G, Lindquist S, Fraenkel E, Yeger-Lotem E.
Nucleic Acids Research 2011 39:W1:W424-9. doi:https://doi.org/10.1093/nar/gkr359
Cellular response to stimuli is typically complex and involves both regulatory and metabolic processes. Large-scale experimental efforts to identify components of these processes often comprise of genetic screening and transcriptomic profiling assays. We previously established that in yeast genetic screens tend to identify response regulators, while transcriptomic profiling assays tend to identify components of metabolic processes. ResponseNet is a network-optimization approach that integrates the results from these assays with data of known molecular interactions. Specifically, ResponseNet identifies a high-probability sub-network, composed of signaling and regulatory molecular interaction paths, through which putative response regulators may lead to the measured transcriptomic changes. Computationally, this is achieved by formulating a minimum-cost flow optimization problem and solving it efficiently using linear programming tools. The ResponseNet web server offers a simple interface for applying ResponseNet. Users can upload weighted lists of proteins and genes and obtain a sparse, weighted, molecular interaction sub-network connecting their data. The predicted sub-network and its gene ontology enrichment analysis are presented graphically or as text. Consequently, the ResponseNet web server enables researchers that were previously limited to separate analysis of their distinct, large-scale experiments, to meaningfully integrate their data and substantially expand their understanding of the underlying cellular response. ResponseNet is available at http://bioinfo.bgu.ac.il/respnet .
2010

Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models.
Su LJ, Auluck PK, Outeiro TF, Yeger-Lotem E, Kritzer JA, Tardiff DF, Strathearn KE, Liu F, Cao S, Hamamichi S, Hill KJ, Caldwell KA, Bell GW, Fraenkel E, Cooper AA, Caldwell GA, McCaffery JM, Rochet JC, Lindquist S.
Disease Models & Mechanisms 2010 3:194-208. doi:https://doi.org/10.1242/dmm.004267
α-Synuclein (α-syn) is a small lipid-binding protein involved in vesicle trafficking whose function is poorly characterized. It is of great interest to human biology and medicine because α-syn dysfunction is associated with several neurodegenerative disorders, including Parkinson’s disease (PD). We previously created a yeast model of α-syn pathobiology, which established vesicle trafficking as a process that is particularly sensitive to α-syn expression. We also uncovered a core group of proteins with diverse activities related to α-syn toxicity that is conserved from yeast to mammalian neurons. Here, we report that a yeast strain expressing a somewhat higher level of α-syn also exhibits strong defects in mitochondrial function. Unlike our previous strain, genetic suppression of endoplasmic reticulum (ER)-to-Golgi trafficking alone does not suppress α-syn toxicity in this strain. In an effort to identify individual compounds that could simultaneously rescue these apparently disparate pathological effects of α-syn, we screened a library of 115,000 compounds. We identified a class of small molecules that reduced α-syn toxicity at micromolar concentrations in this higher toxicity strain. These compounds reduced the formation of α-syn foci, re-established ER-to-Golgi trafficking and ameliorated α-syn-mediated damage to mitochondria. They also corrected the toxicity of α-syn in nematode neurons and in primary rat neuronal midbrain cultures. Remarkably, the compounds also protected neurons against rotenone-induced toxicity, which has been used to model the mitochondrial defects associated with PD in humans. That single compounds are capable of rescuing the diverse toxicities of α-syn in yeast and neurons suggests that they are acting on deeply rooted biological processes that connect these toxicities and have been conserved for a billion years of eukaryotic evolution. Thus, it seems possible to develop novel therapeutic strategies to simultaneously target the multiple pathological features of PD.
2009

Bridging high-throughput genetic and transcriptional data reveals cellular responses to alpha-synuclein toxicity.
Yeger-Lotem E, Riva L, Su LJ, Gitler AD, Cashikar AG, King OD, Auluck PK, Geddie ML, Valastyan JS, Karger DR, Lindquist S, Fraenkel E.
Nature Genetics 2009 41:316-323
Cells respond to stimuli by changes in various processes, including signaling pathways and gene expression. Efforts to identify components of these responses increasingly depend on mRNA profiling and genetic library screens. By comparing the results of these two assays across various stimuli, we found that genetic screens tend to identify response regulators, whereas mRNA profiling frequently detects metabolic responses. We developed an integrative approach that bridges the gap between these data using known molecular interactions, thus highlighting major response pathways. We used this approach to reveal cellular pathways responding to the toxicity of alpha-synuclein, a protein implicated in several neurodegenerative disorders including Parkinson’s disease. For this we screened an established yeast model to identify genes that when overexpressed alter alpha-synuclein toxicity. Bridging these data and data from mRNA profiling provided functional explanations for many of these genes and identified previously unknown relations between alpha-synuclein toxicity and basic cellular pathways.
2005

Alignment of metabolic pathways
Pinter RY, Rokhlenko O, Yeger-Lotem E, Ziv-Ukelson M.
Bioinformatics 2005 21:16:3401-3408. doi:https://doi.org/10.1093/bioinformatics/bti554
Motivation: Several genome-scale efforts are underway to reconstruct metabolic networks for a variety of organisms. As the resulting data accumulates, the need for analysis tools increases. A notable requirement is a pathway alignment finder that enables both the detection of conserved metabolic pathways among different species as well as divergent metabolic pathways within a species. When comparing two pathways, the tool should be powerful enough to take into account both the pathway topology as well as the nodes’ labels (e.g. the enzymes they denote), and allow flexibility by matching similar—rather than identical—pathways.
Results: MetaPathwayHunter is a pathway alignment tool that, given a query pathway and a collection of pathways, finds and reports all approximate occurrences of the query in the collection, ranked by similarity and statistical significance. It is based on a novel, efficient graph matching algorithm that extends the functionality of known techniques. The program also supports a visualization interface with which the alignment of two homologous pathways can be graphically displayed.
We employed this tool to study the similarities and differences in the metabolic networks of the bacterium Escherichia coli and the yeast Saccharomyces cerevisiae, as represented in highly curated databases. We reaffirmed that most known metabolic pathways common to both the species are conserved. Furthermore, we discovered a few intriguing relationships between pathways that provide insight into the evolution of metabolic pathways. We conclude with a description of biologically meaningful meta-queries, demonstrating the power and flexibility of our new tool in the analysis of metabolic pathways.
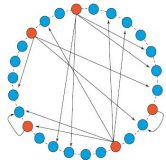
Chromosomal organization is shaped by the transcription regulatory network
Hershberg R, Yeger-Lotem E, Margalit H.
Trends in Genetics 2005 21:3:P138-142. doi:https://doi.org/10.1016/j.tig.2005.01.003
Transcription regulation, a key step in the control of gene expression, has been the focus of several large-scale studies; however, little attention has been given to its relationship with the chromosomal arrangement of transcription units. We studied this relationship systematically in Escherichia coli and Saccharomyces cerevisiae using network analysis methods. Our analysis reveals links between transcription regulation and chromosomal organization, suggesting that in both organisms transcription regulation has shaped the organization of transcription units on the chromosome. Differences found between the organisms reflect the inherent differences in transcription regulation between prokaryotes and eukaryotes.
2004
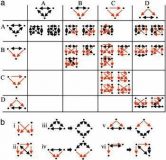
Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction
Yeger-Lotem E, Sattath S, Kashtan N, Itzkovitz S, Milo R, Pinter RY, Alon U, Margalit H
PNAS 2004 101:16:5934-5939. doi:https://doi.org/10.1073/pnas.0306752101
Genes and proteins generate molecular circuitry that enables the cell to process information and respond to stimuli. A major challenge is to identify characteristic patterns in this network of interactions that may shed light on basic cellular mechanisms. Previous studies have analyzed aspects of this network, concentrating on either transcription-regulation or protein-protein interactions. Here we search for composite network motifs: characteristic network patterns consisting of both transcription-regulation and protein-protein interactions that recur significantly more often than in random networks. To this end we developed algorithms for detecting motifs in networks with two or more types of interactions and applied them to an integrated data set of protein-protein interactions and transcription regulation in Saccharomyces cerevisiae. We found a two-protein mixed-feedback loop motif, five types of three-protein motifs exhibiting coregulation and complex formation, and many motifs involving four proteins. Virtually all four-protein motifs consisted of combinations of smaller motifs. This study presents a basic framework for detecting the building blocks of networks with multiple types of interactions.
2003
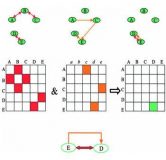
Detection of regulatory circuits by integrating the cellular networks of protein-protein interactions and transcription regulation
Yeger-Lotem E, Margalit H.
The post‐genomic era is marked by huge amounts of data generated by large‐scale functional genomic and proteomic experiments. A major challenge is to integrate the various types of genome‐scale information in order to reveal the intra‐ and inter‐ relationships between genes and proteins that constitute a living cell. Here we present a novel application of classical graph algorithms to integrate the cellular networks of protein–protein interactions and transcription regulation. We demonstrate how integration of these two networks enables the discovery of simple as well as complex regulatory circuits that involve both protein–protein and protein–DNA interactions. These circuits may serve for positive or negative feedback mechanisms. By applying our approach to data from the yeast Saccharomyces cerevisiae, we were able to identify known simple and complex regulatory circuits and to discover many putative circuits, whose biological relevance has been assessed using various types of experimental data. The newly identified relations provide new insight into the processes that take place in the cell, insight that could not be gained by analyzing each type of data independently. The computational scheme that we propose may be used to integrate additional functional genomic and proteomic data and to reveal other types of relations, in yeast as well as in higher organisms.
1999
The AS/400 cluster engine: A case study
Gera Goft, Esti Yeger-Lotem
In this paper we share our experience in harnessing group communications for the AS/400 cluster infrastructure. The main fonts of the AS/400 cluster is providing high availability and disaster recovery of defined cluster resources. The cluster supports up to 128 nodes, connected via any IP network. Cluster nodes and resources can be dynamically added or removed. Administrative actions or failures trigger automatic cluster reconfigurations: CLUE (Cluster Engine) is the group communication middleware implemented in the OS/400 kernel. It exploits the strong virtual synchrony model. CLUE is unique in the following aspects: It is the first virtually synchronous group communication system incorporated into a cluster solution, and customized to meet cluster needs. It is the only group communication system (GCS) integrated into a commercial operating system kernel. CLUE’s special features (e.g., Flexible Group Member) can be relevant to other group communication systems.